


UC
-
Posts
547 -
Joined
-
Last visited
Content Type
Profiles
Forums
Events
Everything posted by UC
-
That particular power of HF is all about fluoride and it's affinity for silicon. Since all the acids do different things to different substrates, I don't think it's really fair to pick based on reactions of the anion. The only fair grounds to compare acids is the acidity and the acidity alone (what you are saying). The pKa for HeH+ is entirely theoretical, by the way. There is no way that compound could ever be isolated and used in a lab. (perhaps a few molecules in some fancy physics equipment at most)
-
Well, this is also just a guess, but aside from what swansont said, wouldn't water molecules along crystal grain boundaries and in crystal defects be slightly higher energy than those in regions of perfect crystallinity? Rate of sublimation would therefore be slightly higher along the crystal boundaries (which are extremely numerous unless you go to great lengths to freeze high purity water very slowly).
-
either this guy is incredibly thick or he's a subtle troll who's rolling around on the floor laughing right now.
-
Look up the hammet acidity function. In determining acidity, regardless of solvent, what matters is how strongly the acidic hydrogen is bonded to the rest of the acid. Believe it or not, water is quite capable of behaving as a base. You will never see free [ce] H^+ [/ce] ions in water to any appreciable degree because free protons would be the strongest possible acids by definition. Instead, you have a reaction with the water, forming the hydronium ion, [ce] H3O^+ [/ce], which is the strongest acid that can exist in aqueous solution. Any acids that are stronger than hydronium ion will effectively completely react with water to give hydronium ion and an anion. [ce] HX + H2O -> H3O^+ + X^- [/ce] A 0.01F solution of [ce] HNO3 [/ce] and a 0.01F solution of [ce] HBr [/ce] in water will have effectively the same pH. In other solvents, this will not be the If you want to consider how dangerous things are, you need to consider a whole slew of other characteristics. The ability to concentrate acids is important. concentrated sulfuric, for example, is a very powerful dehydrating agent and has a very strong heat of hydration (add water and it gets very very hot), which makes spills extremely dangerous. Hydrofluoric acid, since it reacts will all sorts of things, and is extremely toxic to life, has it's own dangers even though it is a weaker acid in aqueous solution than many other acids. Nitric acid is a very powerful oxidizer, so like hermann said, it will put holes in many metals or react violently with organic compounds (and release clouds of toxic gas in both cases). These same metals might be completely immune to concentrated sulfuric or hydrochloric acid.
-
Porous ice would be very good at adsorbing smells, not absorbing them
-
You won't get any apples during the spring either. They're a fall crop. I would sit very still and quietly and wait for the non-zero probability event to occur where my entire body would simultaneously and concertedly tunnel to the other side of the river, allowing me to get the apples
-
I edited some of my text while you were typing that. Sorry I'm at home for the summer and scifinder is dead as a doornail off-campus as well.
-
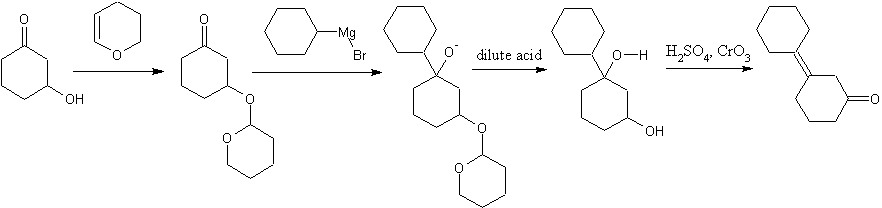
Well, you can't argue with published procedure. I do not have access to those resources, but I'll take your word for it. Since I am in college and am heading for a career in organic synthesis, would you mind running through a general procedure for monoprotection, in PMs if you'd prefer. Do you use 1eq of the alcohol in an inert solvent with acid catalysis? I rather like to have a realistic procedure, even if it is just a theoretical exercise. It's good practice. Instead of THP, how about trapping one of the ketone functionalities as an enol ether with TSMCl? Or will that be unstable to grignard reagents? a dash of fluoride in the acidic workup and any diastereomer related problems go away.
-
You will not get allylic bromination on cyclohexanone. Ketones are rapidly halogenated in the alpha-positions by an electrophilic reaction involving the enol. Allylic brominations are usually done with N-bromosuccinimide in nonpolar solvent, where you will generate precisely none of the enol. Who's to say what side of the enol the bromine would attack anyway. You would get a mix of 2 and 3-bromocyclohexanones if it worked. Also, you have the hydrolysis of a secondary alkyl halide with NaOH. If you've covered the E1,E2, SN1, and SN2 mechanisms, you know that secondary alkyl halides are fair game for just about any reaction mechanism, so you will probably get some elimination as well. I'm pretty sure that you will get borane addition across both bonds, not just the one, adding an extra dehydration step. You can come into chat if you'd like to discuss in person. Hit the "chat" button on the top menu bar. Are you required to start with cyclohexanol?
-
You won't be able to efficiently monoprotect one of the two ketones. Protection is done in an excess of alcohol with acid catalyst to ensure it goes to completion. An ether protected 3-hydroxycyclohexanone is your best option, although using PCC to oxidize an alcohol is a waste unless acidified dichromate or another cheapo oxidizer won't work. PCC has the lovely distinction of having both pyridine, hexavalent chromium, and is typically used in dichloromethane. Acidified dichromate may have the hexavalent chromium, but has no need for pyridine or toxic organic solvent.
-
*cries in his soup* How will I ever get by without that annoying noise I ocasionally hear in the chatroom!?!?!
-
the field would be effectively equivalent in all directions, and the net field would be 0.
-
Nickel!?!?
-
Keep in mind that conversion of tertiary alcohols is usually done in concentrated HX, so there is quite a bit of hydronium floating around. Consider also that the tertiary halide is insoluble in water, so it seperates out and removes itself from the reaction. tertiary alcohol + HX <--> tertiary alkyl oxonium halide <--> carbocation + water + halide ion --> tertiary alkyl halide + water
-
I sent my prof. an email as well. I have no idea when he'll respond since semester is over. I kind of get the feeling that nobody cares and since the -dig describes the whole reaction, not just a step, it would be presferred.
-
Well, based on the very short lived intermediate, it would be -trig, but if you consider only the starting material and the ending material, -dig is appropriate. I'm not sure which is technically correct.
-
By a very long shot, the easiest way to make copper salts is to buy a lot of copper sulfate from somewhere and work from that. If you're really desperate, bubbling air through a solution of ammonium sulfate in aqueous ammonia with copper metal sitting in it is pretty good at oxidizing the copper and dissolving it as tetraamminecopper hydroxide. From there, drying out the solution and heating to decompose the complex and drive off ammonia, then dissolving in your acid of choice is a decent, but very annoying way to go.
-
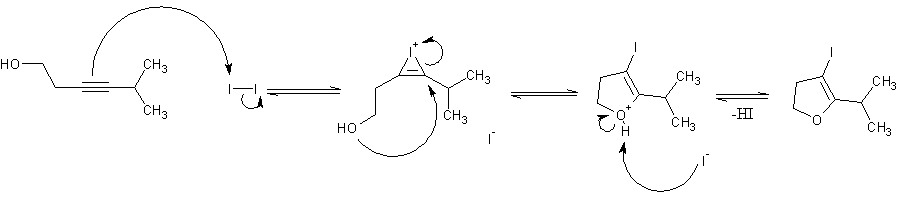
Funny. I'd never even heard of Baldwin's rules until now. I suspect that if you were to move the alkyne one carbon closer to the alcohol group and throw an alkyl chain on the other end of it, you could get 5-endo-dig cyclicization via a similar mechanism, giving a 3-iodo-2,3-dihydrofuran ring.
-
how would you ionize hydrogen atoms to get just protons?
UC replied to cameron marical's topic in Quantum Theory
This will exclusively make hydronium ions [ce] H3O^+ [/ce]. [ce] H^+ [/ce] is just shorthand, since the net effect is the motion of a proton, but it occurs exclusively as hydronium in aqueous solution, or appropriate species in other solvents. The closest you can get to naked protons in a reaction mixture is 1:1 molar combination of antimony pentafluoride and liquid anhydrous hydrofluoric acid. This is the strongest known superacid. Swansont is the guy to listen to here. -
Or miniature models of the backstreet boys and N'sync.
-
The only way you'll be able to grow basic copper carbonate crystals is maybe in a pressurized high temperature tube with a temperature gradient and the solution saturated with carbon dioxide or bicarbonate. Everyone thinks electrolysis is some sort of magic bullet. It's not.
-
what is this crap? absolute detritus.
-
That you're on one hell of a drug trip.
-
Potassium manganate --> Potassium permanganate HELP
UC replied to jerryshizzle123's topic in Inorganic Chemistry
Hrm. Now that I think about it, nevermind. Chlorine won't work anyway. The oxidizing potential of permanganate is too high. If you make the solution acidic, [ce] Cl^- [/ce] will take it all the way down to [ce] Mn^2^+ [/ce]. [ce] MnO2 [/ce] with hydrochloric acid was an old way to generate chlorine gas. in basic solution, it will remain as manganate. -
Potassium manganate --> Potassium permanganate HELP
UC replied to jerryshizzle123's topic in Inorganic Chemistry
The fact that you don't know means you shouldn't even think about it. Just read the wiki article. I suspect that has enough info.