


Scienc
-
Posts
18 -
Joined
Content Type
Profiles
Forums
Events
Posts posted by Scienc
-
-
I have a question about the value obtained by Rate Law. For example, in this case:
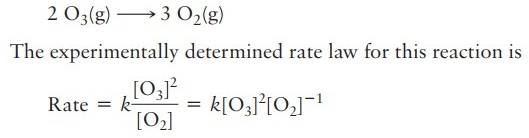
the value of rate obtained by this equation is the global rate of reaction, right, and not the rate of a specific specie, I know that using this global rate I can find the species rate, multiplying for his stoichiometric numbers.
Is my assumption correct? Because if the answer is YES, this question no make sense for me.
I was trying to solve this question:

I created this graph (Title is in Portuguese):
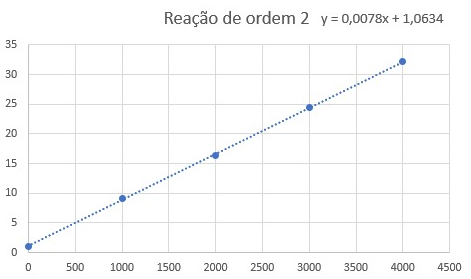
I used "1/[HI] x t," which indicates a second-order reaction with a rate law equal V = k[HI]^2, so, the slope of the graph is numeric equal for k, AND, if my previous assumption is correct, the value obtained for V is the global rate reaction, as a consequence, the constant (k) 0.0078 (L.mol-1s-1) represent the unique rate law and not of specific reactant (HI).
However, the gab, says that 0.0078 represents the rate constant (K) for the HI
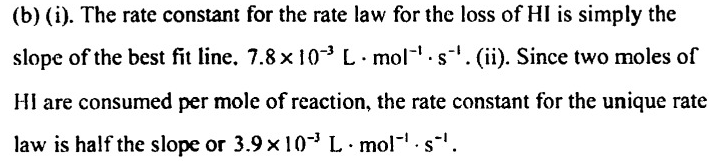
For me, it makes no sense, because, if it is correct, the value of V obtained by rate laws is the rate of HI and not for the global reaction since k refers to him and not for the global reaction.
0 -
1 hour ago, exchemist said:
I don't know the first thing about this program, but it sounds to me as if it may use some sort of method of iterative approximation that converges towards a final value. If that is so it may be programmed to stop iterating when the difference between successive iterations falls below a preset level, or something. Could that makes sense?
Thanks.
1 hour ago, StringJunky said:Thank so much,
1 -
I am a beginner in QE and I have a question. During I was reading about Output file, I saw a part where have it:
"The calculation goes on step by step until convergence is reached. Then we show all the energies of all bands (eigenvalues of the Kohn-Shan orbitals) for each k-point, the Fermi energy, the total energy, the contributions of all the terms to the total energy, and the calculation time used by each subroutine of the program."
Output file:
... iteration # 9 ecut= 100.00 Ry beta=0.70 Davidson diagonalization with overlap ethr = 1.00E-13, avg # of iterations = 1.0 negative rho (up, down): 1.151E-06 0.000E+00 total cpu time spent up to now is 28.4 secs End of self-consistent calculation k = 0.0000 0.0000 0.0000 ( 3909 PWs) bands (ev): -17.8569 -5.9595 -1.3957 -1.3957 4.4242 9.2594 9.9561 9.9561 k = 0.0000 0.0550 0.0000 ( 3909 PWs) bands (ev): -17.8034 -5.8955 -1.6155 -1.5115 4.4968 9.3266 9.9837 10.1886 ... -10.9411 -10.9411 -8.9101 1.6562 1.6562 12.4084 14.2298 14.2298 the Fermi energy is 1.6562 ev ! total energy = -22.80493136 Ry Harris-Foulkes estimate = -22.80493136 Ry estimated scf accuracy < 1.5E-15 Ry The total energy is the sum of the following terms: one-electron contribution = -44.02527202 Ry hartree contribution = 24.36506286 Ry xc contribution = -6.99363523 Ry ewald contribution = 3.85762303 Ry Dispersion Correction = -0.00870488 Ry smearing contrib. (-TS) = -0.00000512 Ry convergence has been achieved in 9 iterations Writing output data file grafeno.save init_run : 2.26s CPU 2.29s WALL ( 1 calls) electrons : 20.10s CPU 20.24s WALL ( 1 calls) ... Parallel routines fft_scatter : 4.35s CPU 4.44s WALL ( 23020 calls) PWSCF : 28.46s CPU 28.70s WALL This run was terminated on: 11:51: 0 5Sep2017 =------------------------------------------------------------------------------= JOB DONE. =------------------------------------------------------------------------------=0 -
6 hours ago, sethoflagos said:
The exercise is misleadingly worded.
If a +ve amount of work is 'done on the system' then this must be understood as energy being added to the system whatever sign convention you are following, and therefore the internal energy change arising from that work must increase. This is where your method gave the wrong answer.
The term 'expansion work' is commonly used as a synonym for any PdV process even when, as in your example, the system is being compressed. We know that the book intends compression because their PdV term is negative, and therefore dV is negative.
So, is the true value for work +22, because work is done on the system?
9 hours ago, joigus said:Think about it. You must provide heat to make the pressure constant during an expansion.Hm. There's something more to it. I can't read the image very well..
Sorry, I misread. It's a chemical reaction.
OK. I think I see what the problem is. The criterion that you use is consistent with w=−PΔV , right?
Not with w=+PΔV
Some people write ΔU=q−w , and other people prefer ΔU=q+w , but when expressed in terms of P, V, in the 1st principle, it must always be, δU=q−pΔV , because if it's an expansion, the contribution to the internal energy must be negative. That's what's written in stone.
Maybe you got confused with a sign criterion you saw somewhere else?
Sorry for the initial confusion. Was that helpful?
I used the 𐤃U = q+w. because I studied in a chemical class. However, I considered the contribution to work as negative. The book on the other hand, used a positive signal for work.
The images is from the book.
0 -
I have a question about the resolution of this exercise:

I tried to solve this problem with the following steps:
For the 1º law of thermodynamics, 𐤃U = q+w.
As q = 𐤃H at constant pressure, I assumed that in this process q = - 15 kJ.
As a result, 𐤃U = -15 - 22 (because was an expansion) and for this reason, 𐤃U = -37 kJ.
However, the book has this resolution

I understood why the book used this method, but I did not understand why my method is wrong.
0 -
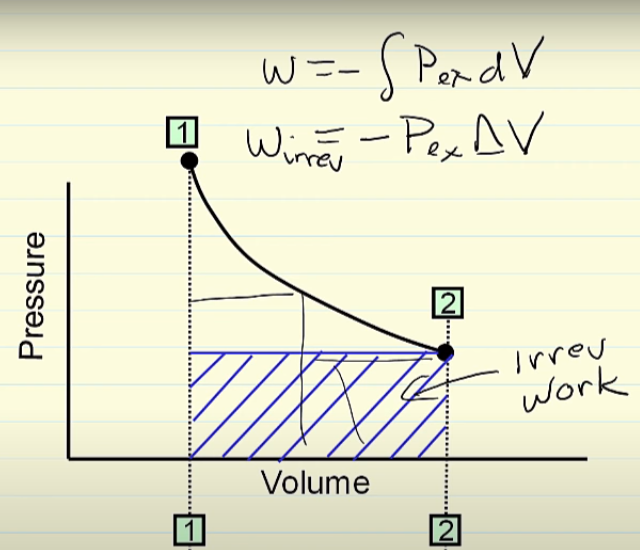
Why in an irreversible process, is the pressure constant? I understood why I should use Wirrev = -PΔV, but I did not understand why the pressure is constant in this process.
0 -
Why does CH3CO2H have a greater nucleophilicity than CH3OH?
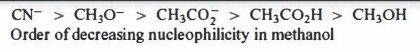 0
0 -
An orbital can contain up to 2 electrons, and the electrons and orbitals are described by wave functions, are the 3 wave functions the same? for example, two electrons that are in the psi 2 0 0 orbital do they have the same wave function as that orbital?
0 -
What does "air is saturated with water" or "air is saturated with something" and how can I relate this to vapor pressure?
0 -
What is the physical meaning of enthalpy?
0 -
16 minutes ago, joigus said:
I think you're confusing change in volume with change in amount of matter present in your system. Moles are a measure of the number of atomic participants in your system. Volume is very different. Please review your statement so that a proper answer can be given to what's troubling you.
The system, such as you've defined it, is an open system (open to exchanges of both energy and matter).
The volume that 6 moles occupy 134.4l and 12 moles occupy 268.8 moles, since 1 mol = 22.4l, for this the gas needs to expand
0 -
What is the difference between the work done by a gas and the workflow that the same gas performs to exist? why does the variation in the internal energy of the equation dH = dQ + nRT not consider the work done by the gas to exist (working flow) but consumes only the heat transfer?
In the question below, the author considers dU = Q. He performs the calculation of the work done by the gas, separated from the internal energy (with an equation dH = dU + nRT ). If you know that in systems with constant volume dU = Q, but the gas volume varies (From 6 moles to 12 moles of gas), this does not imply an internal energy variation equal to dU = w + q (As stated in the 1st law of thermodynamics) and why consider dU = w + q and not dU = Q **? In practice or in what he did, the logic he did not use was.
Question:
“A constant volume calorimeter showed that the fuel loss in burning 1 mole of glucose is equal to -2559kJ in 298K, that is dU = -2555KJ”
0 -
14 minutes ago, joigus said:
Outra maneira de dizer: a entalpia é uma função do estado que permite expressar os balanços de calor. O calor não é uma função do estado, mas você pode relacioná-lo com uma função do estado. Algum tipo de "potencial de calor". Isso não é legal? Você deve manter a pressão constante se quiser que ela faça seu trabalho como "potencial de calor".
Isso ajuda?
Thank you.
4 hours ago, sethoflagos said:A energia interna está principalmente associada ao volume fixo, processos fechados onde dU = CvdT se mantém. A entalpia está associada a processos abertos e de pressão fixa, nos quais dH = CpdT se mantém. Em processos abertos, o fluxo de produtos está sendo executado no espaço já ocupado por outra coisa e essa outra coisa precisa ser movida para outro lugar. O trabalho adicional necessário está contido no termo PV em sua fórmula H = U + PV. Portanto, em experimentos de laboratório de química ou processos industriais, geralmente trabalhamos com alterações de entalpia, em vez de alterações na energia interna. Poderíamos usar o U o tempo todo, mas teríamos que acompanhar todos os termos de PV externos e é fácil perder o um ou dois ímpares.
Existem muitas "rugas" no exposto, mas acredito que você esteja mais interessado na ampla visão geral e, nesse nível, o exposto acima funciona bem para mim. Mais elaboração pode ser encontrada nos livros apropriados.
Thank you.
0 -
A variation of enthalpy at constant pressure is numerically equal to the variation of heat in a chemical reaction, but what is enthalpy in itself? what is its difference for internal energy, is it calculated as H = U + PV, or does that mean physically?
0 -
Guys, why in physics does the work done by the system have the POSITIVE sign, but in chemistry does the work done by the system have the NEGATIVE sign?
0 -
Why does the equation τ = -n.R.T.ln (v_2 / v_1) only work for reversible processes?
0
Quantization of angular momementum
in Homework Help
Posted
Can someone give me a hint on how can I prove it?